课题组导师
刘娟
个人简介
武汉大学二级教授、博士生导师。武汉大学计算机学院人工智能研究所所长,教授委员会副主任。武汉大学首批“351”人才计划珞珈特聘教授,曾入选教育部骨干教师计划、教育部新世纪人才计划。
教育背景
1991获武汉大学计算机系计算机软件专业理学学士学位
1996获计算机科学与理论专业工学博士学位
1999赴香港浸会大学计算机系访问
2000年9月-2002年10月在日本东京大学大学院做博士后研究员
行业服务
目前是中国计算机学会高级会员及生物信息专业组委员,中国人工智能学会高级会员及生物信息及人工生命专委会委员,中国运筹学会会员及系统生物分会理事,中国医疗保健国际交流促进会会员及健康大数据和数字化医疗分会委员,中国妇幼保健协会会员及妇女病防治专业委员会委员,国家科学技术进步奖评审专家、教育部专家库专家、教育部学位与研究生教育评估专家、国家科技专家库专家、湖北省科技奖励评审专家等。
研究方向
生物信息,机器学习、数据挖掘、模式识别、医学图像处理等。已发表相关研究论文150余篇,已获批或申请国家发明专利或软件著作权多项。主持或承担国家自然科学基金、科技部重大仪器专项等多项课题。多个国际会议的组委会成员或PC Member, 多个国内外学术期刊的编委或论文评审人。
冯晶
个人简介
博士,计算机学院副教授,硕士生导师 研究方向为生物信息、模式识别和数据挖掘。主要从事高通量生物大数据的挖掘分析和可视化工作,包括机器学习和深度学习方法在海量生物数据以及医疗数据(特别是心电数据和病理图片数据)中的应用。
代表论文
1. Siyu Xia, Jing Feng, Ke Chen, and et al, CSCD: A Database for Cancer-specific Circular RNAs. Nucleic
Acids Res. 2018 Jan 4; 46(D1): D925-D929 (共同第一作者, SCI IF:11.561)
2. Jing Gong, Shufang Mei, Chunjie Liu, Yu Xiang, Youqiong Ye, Zhao Zhang, Jing Feng, and et al,
PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids
Res. 2018 Jan 4;46(D1): D971-D976. (SCI IF:11.561)
3. Jing Feng, Yu Xiang, Siyu Xia, and et al, CircView: A Visualization and Exploration Tool for
Circular RNAs. Briefings in Bioinformatics. 2017 Jun 30. (SCI IF: 6.302)
4. Lijun Lei, Siyu Xia, Dan Liu, Xiaoqing Li, Jing Feng, Yaqi Zhu, Jun Hu, Linjian Xia, Lieping
Guo, Fei Chen, Hui Cheng, Ke Chen, Hanyang Hu, Xiaohua Chen, Feng Li, Shan Zhong, Nupur Mittal, Guohua
Yang, Zhijian Qian, Leng Han and Chunjiang He: Genome-wide Characterization of LncRNAs in Acute Myeloid
Leukemia. Briefings in Bioinformatics. 2017. (SCI IF: 6.302)
5. Siyu Xia, Jing Feng, Lijun Lei,and et al, Comprehensive Characterization of Tissue-specific
Circrular RNAs in the Human and Mouse Genomes. Briefings in Bioinformatics. 2016. (共同第一作者, SCI IF:
6.302)
6. Chunjiang He, Hanyang Hu, Kitchener D. Wilson, Haidi Wu, Jing Feng, Siyu Xia, Jared Churko,
Kun Qu, Howard Y. Chang and Joseph C. Wu: Systematic Characterization of Long Non-Coding RNAs Reveals
the Contrasting Coordination of Cis- and Trans- Molecular Regulation in Human Fetal and Adult Heart:
CIRCGENETICS. February 2016. (SCI)
7. Limin Zhou, Wei Zheng, Majing Luo, Jing Feng, Zhichun Jin, Yan Wang, Dunlan Zhang: dbCerEx -
A web-based database for the analysis of cervical cancer transcriptomes. PLOS one. 2014 9(6): 1-5.
(SCI).
主要成果
1. 肿瘤特异环状RNA数据库
对来自19种癌症的细胞系进行了癌症特异的环状RNA筛选,预测其MRE及RBP结合位点,以及ORF阅读框,并对母本基因的选择性剪接进行了定义。
http://gb.whu.edu.cn/CSCD
2.组织特异环状RNA数据库
对超过16种人和小鼠组织的环状RNA进行了特异性识别和定位,并预测了其MRE及RBP结合潜能。
http://gb.whu.edu.cn/TSCD
3.环状RNA可视化工具
可用于对组织与疾病特异环状RNA进行比对、识别和筛选,以及预测其MRE和RBP结合,为实验验证环状RNA提供原始信息。
http://gb.whu.edu.cn/CircView
https://github.com/GeneFeng/CircView
张丽华
个人简介
武汉大学计算机学院副研究员,硕士生导师,主要从事生物信息学、人工智能与大数据方面的研究,主要围绕最优化理论、模式识别、统计机器学习等方法,开展(单细胞)生物医学大数据整合挖掘分析的模型与算法研究。主要成果发表在Genome Biology, Nucleic Acids Research, IEEE Transactions on Fuzzy Systems 等知名学术期刊。
教育背景
2013年9月至2018年6月在中国科学院数学与系统科学研究院硕博连读
2018年8月至2021年7月在美国加州大学尔湾分校从事博士后研究
2021年9月加入武汉大学计算机学院
代表论文
1.Lihua Zhang, Qing Nie. scMC learns biological variation through the alignment of
multiple single cell
genomics datasets. Genome Biology. 2021, 22(1):10.
2.Suoqin Jin*,Lihua Zhang*(共同一作), Qing Nie. scAI: an unsupervised approach for
integrative analysis of parallel single cell transcriptomic and epigenomic profiles. Genome
Biology.2020, 21(1):25.
3.Lihua Zhang, Shihua Zhang. Learning common and specific patterns from data of
multiple interrelated biological scenarios with matrix factorization. Nucleic Acids
Research.
2019, 47(13): 6606-6617.
4.Lihua Zhang, Shihua Zhang. A general joint matrix factorization framework for data
integration and its systematic algorithmic exploration. IEEE Transactions on Fuzzy
Systems. 2019, 28(9): 1971-1983.
5.Lihua Zhang,Shihua Zhang. Imputing single-cell RNA-seq data by considering cell
heterogeneity and prior expression of dropouts. Journal of Molecular Cell Biology.
2020, doi:10.1093/jmcb/mjaa052.
6.Lihua Zhang, Shihua Zhang. Comparison of computational methods for imputing
single-cell RNA-sequencing data.IEEE/ACM Transactions on Computational Biology and
Bioinformatics. 2020, 17(2): 376-389.
7.Suoqin Jin, Christian Guerrero-Juarez,Lihua Zhang, Ivan Chang, Raul Ramos,
Chen-Hsiang Kuan, Peggy Myung, Maksim V. Plikus, Qing Nie. Inference and analysis of cell-cell
communication using CellChat. Nature Communications. 2021, 12(1088).
8.Laiyi Fu, Lihua Zhang, Emmanuel Dollinger, Qinke Peng, Qing Nie#, Xiaohui Xie#.
Predicting transcription factor binding in single cells through deep learning. Science
Advances. 2020, 6(51): eaba9031.
研究成果
主要从事单细胞等生物医学大数据挖掘和机器学习的模型与算法研究,并与生物医学家紧密合作解决前沿科学问题。具体包括以下四个方面:
1. 单细胞转录组、表观组等多组学数据的挖掘与整合建模及其算法设计
- 跨平台、跨物种、不同实验条件下的单细胞多组学数据整合
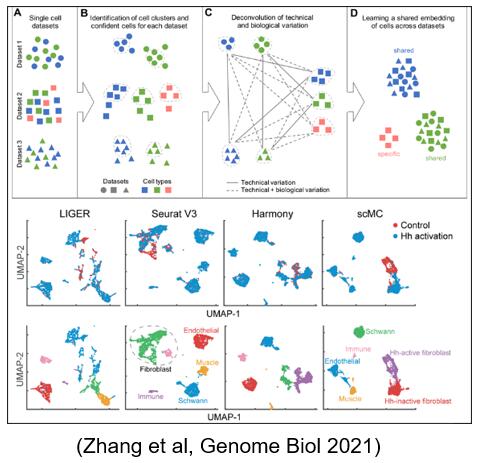
基于方差分析和优化技术,提出一个整合多种条件下单细胞转录组或表观组生物数据的优化模型与算法scMC,首次提出通过区分生物和技术噪声来防止数据的过整合。与主流的单细胞数据整合方法Seurat, Harmony, LIGER相比,scMC能够更为精确地整合单细胞数据,挖掘潜在的共同和差异的细胞亚群, 尤其是来源于不同生物条件下的数据。
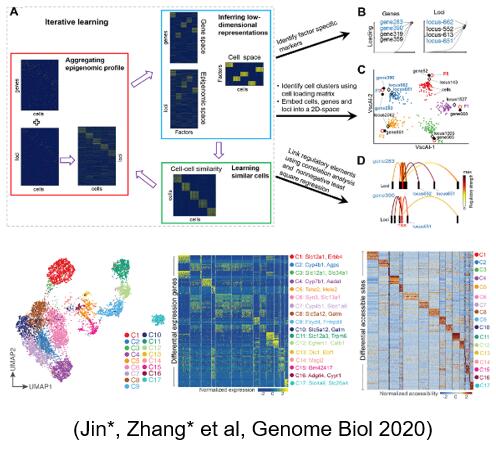
针对单细胞表观组数据的稀疏与二值化特性,创新性地提出基于优化技术自适应增强稀疏信号的方法scAI,并通过迭代学习框架将其与单细胞转录组数据进行整合降维和挖掘分析,为单细胞多组学数据的整合分析提供了高效的计算方法。利用scAI,揭示出基因表达谱相近的细胞亚群之间存在着明显的染色质开放程度上的差异。
- 单细胞高通量测序数据填补去噪
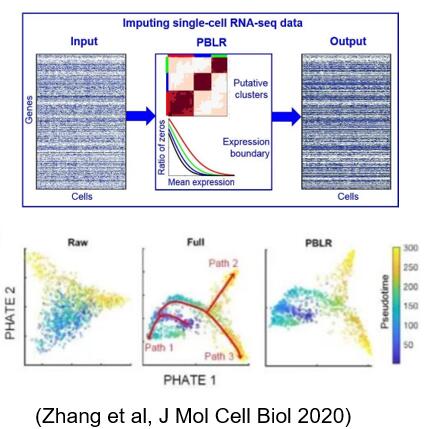
充分利用单细胞转录组数据的分布特点,提出基于细胞子群和基因表达缺失先验信息的单细胞转录组测序数据填补的方法PBLR,有效地解决数据缺失及稀疏的问题、提升单细胞转录组数据分析的精确性。与主流的6种单细胞数据填补方法相比, PBLR能够更为精确地恢复缺失的数据,能有效提升数据低维可视化、提取基因间关系等下游分析能力。
- 空间转录组数据的建模与分析
- 基因调控网络的推断和分析
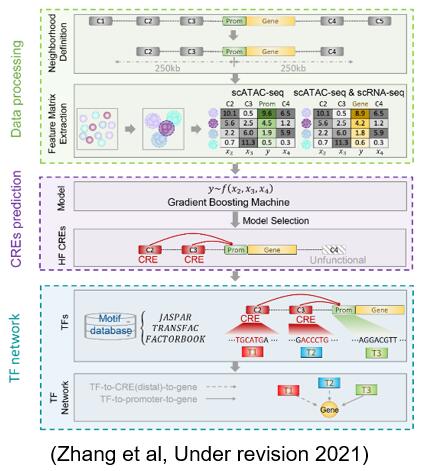
单细胞染色质开放程度捕获技术的发展,为从单个细胞水平研究转录调控机制提供了机会。提出整合单细胞转录组和染色质开放程度数据,识别强功能性的转录调控元件,并构造细胞子群特异的转录调控元件的工具包。通过该方法发现了脑区中两个细胞亚型对应的差异调控元件及其调控关系网络,指出其与抑郁症相关的突变基因的联系。
- 细胞通讯网络的推断和分析
- 癌症等疾病的基因组、转录组、表观组等分子层面的变异模式
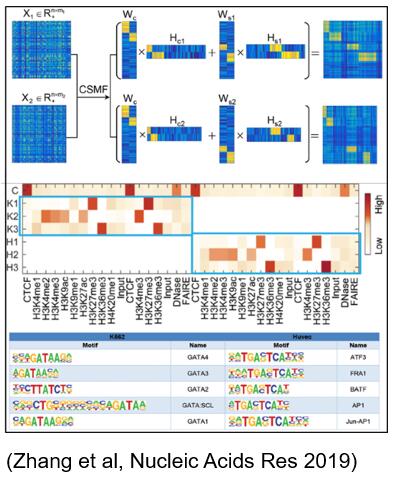
高通量测序技术产生了大规模的相互关联的生物医学组学数据如不同癌症、不同时刻等,如何对这些相互关联的组学数据进行模式识别和差异分析是一个重要且具有挑战性的问题。提出了第一个同时识别共有和特异组合模式的优化模型,该模型基于联合非负矩阵分解技术和创新的参数估计方法,具有很强的可解释性,便于从相关生物情景的数据中提取具有显著生物学意义的共有和特异模式
- 转录调控元件的功能(促进、抑制、组合)预测
- 分子特征层面到表型的因果推断
- 与生物医学家紧密合作,研究白癜风的致病机制
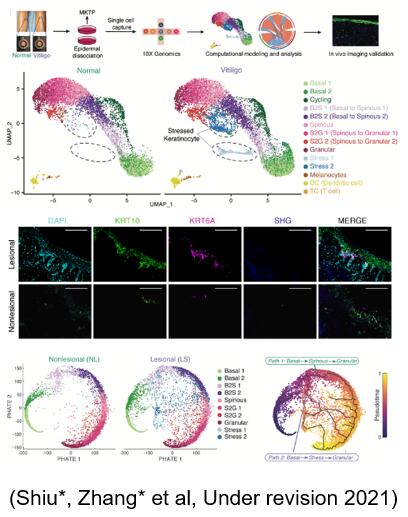
将开发的单细胞数据整合算法工具scMC,与医学家合作,揭示了白癜风的高度异质性以及发现致病信号通路。在白癜风样本中检测到特异的stressed Keratinocytes 细胞,这类细胞表现出氧化磷酸化、糖酵解等代谢方面的差异。Keratinocytes细胞分化过程对比正常分化过程外,白癜风样本中还存在一条经过Stress keratinocytes细胞的分化路径,在细胞相互作用分析中发现其与免疫细胞之间的交流差异,这些计算分析的结果进一步得到了实验验证。
田天
个人简介
田天教授专注于生物大数据的人工智能算法,围绕生物组学数据中的高噪音、高维度、多组学融合、批次多样性等计算挑战为关键科学问题,开发创新人工智能算法,取得了具有一定影响力的学术成绩。近年来共发表SCI学术论文30余篇,其中以第一作者在生物信息学国际主流学术期刊发表研究论文11篇, 包括 Nature Methods、Nature Machine Intelligence、 Nature Communications、Genome Research、EBioMedicine等。合作成果发表于Nature Medicine、Cancer Discovery、PNAS等。担任Frontiers in Genetics和BMC Bioinformatics编辑。单篇研究性论文最高引用220余次。获得 2023 年国家海外高层次人才青年项目。
代表论文
1. T. Tian, J. Wan, Q. Song, Z. Wei, Clustering Single-cell RNA-seq Data with A Model-based Deep Learning Approach, Nature Machine Intelligence (2019) 1 (4), 191–198.
2. T. Tian, J. Zhang, X. Lin, Z. Wei, H. Hakonarson. Model-based Deep Embedding for Constrained Clustering Analysis of Single-cell RNA-seq Data. Nature Communications (2021) 12 (1), 1-12.
3. T. Tian, Z. Wei, X. Chang, Y. Liu, RE Gur, PMA Sleiman, H. Hakonarson, The Long Noncoding RNA Landscape in Amygdala Tissues from Schizophrenia Patients, EBioMedicine (2018) 34, 171181
4. T. Tian, C. Zhong, X. Lin, Z. Wei, H. Hakonarson. Complex hierarchical structures in single-cell genomics data unveiled by deep hyperbolic manifold learning. Genome Research (2023) 33 (2), 232-246.
5. T. Tian, J. Zhang, X. Lin, Z. Wei, H. Hakonarson. Dependency-aware deep generative models for multitasking analysis of spatial genomics data. (accepted by Nature Methods)
6. F. Tan *, T. Tian* , X. Hou, X. Yu, L. Gu, F. Mafra, B. Gregory, Z. Wei, H. Hakonarson, Elucidation of DNA Methylation on N6 -Adenine with Deep Learning, Nature Machine Intelligence (2020) 2 (8), 466-475. (* co-first authors).
7. L. Xiang* , T. Tian* , Z. Wei, H. Hakonarson. Clustering of single-cell multi-omics data with a multimodal deep learning method. Nature Communications (2022) 13(1), 1-18. (* co-first authors).