Professor and doctoral tutor at Wuhan University. Director of the Institute of Artificial Intelligence, School of Computer Science, Wuhan University, deputy director of the Professor Committee. Wuhan University's first batch of "351" talent plan "LuoJia special professor", was selected as the backbone of the Ministry of Education teacher program, the Ministry of Education New Century Talent Program.
In 1991, obtained a bachelor's degree in computer software from Wuhan University
In 1996, obtained a doctorate in computer science and theory
In 1999, went to the Computer Department of Hong Kong Baptist
University
From September 2000 to October 2002, he worked as a postdoctoral researcher at the University
of Tokyo, Japan
She is currently a senior member of the Chinese Computer Society and a member of the Bioinformatics Professional Group, a senior member of the Chinese Artificial Intelligence Society, a member of the Bioinformatics and Artificial Life Committee, a member of the China Operations Research Society and a member of the System Biology Branch, and a member of the China Healthcare International Exchange Promotion Association. Member of Health Big Data and Digital Medical Branch, member of China Maternal and Child Health Association and member of Women's Disease Prevention and Treatment Committee, National Science and Technology Progress Award Evaluation Expert, Ministry of Education Expert Library Expert, Ministry of Education Degree and Postgraduate Education Evaluation Expert, National Science and Technology Expert Library Expert, Hubei Science and Technology Awards Evaluation Experts.
Biological information, machine learning, data mining, pattern recognition, medical image processing, etc. He has published more than 150 related research papers and has been approved or applied for a number of national invention patents or software copyrights. Host or undertake a number of topics such as the National Natural Science Foundation of China and the major instrument specialties of the Ministry of Science and Technology. Member of the organizing committee of several international conferences or PC Member, editorial board of several domestic and foreign academic journals or reviewers of papers.
Ph.D., Associate Professor, School of Computer Science, Master's Tutor His research interests include bioinformatics, pattern recognition and data mining. Mainly engaged in the mining analysis and visualization of high-throughput biological big data, including the application of machine learning and deep learning methods in massive biological data and medical data (especially ECG data and pathological image data).
1. Siyu Xia, Jing Feng, Ke Chen, and et al, CSCD: A Database for Cancer-specific Circular RNAs. Nucleic
Acids Res. 2018 Jan 4; 46(D1): D925-D929 (co-author, SCI IF:11.561)
2. Jing Gong, Shufang Mei, Chunjie Liu, Yu Xiang, Youqiong Ye, Zhao Zhang, Jing Feng, and et al,
PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids
Res. 2018 Jan 4;46(D1): D971-D976. (SCI IF:11.561)
3. Jing Feng, Yu Xiang, Siyu Xia, and et al, CircView: A Visualization and Exploration Tool for
Circular RNAs. Briefings in Bioinformatics. 2017 Jun 30. (SCI IF: 6.302)
4. Lijun Lei, Siyu Xia, Dan Liu, Xiaoqing Li, Jing Feng, Yaqi Zhu, Jun Hu, Linjian Xia, Lieping
Guo, Fei Chen, Hui Cheng, Ke Chen, Hanyang Hu, Xiaohua Chen, Feng Li, Shan Zhong, Nupur Mittal, Guohua
Yang, Zhijian Qian, Leng Han and Chunjiang He: Genome-wide Characterization of LncRNAs in Acute Myeloid
Leukemia. Briefings in Bioinformatics. 2017. (SCI IF: 6.302)
5. Siyu Xia, Jing Feng, Lijun Lei,and et al, Comprehensive Characterization of Tissue-specific
Circrular RNAs in the Human and Mouse Genomes. Briefings in Bioinformatics. 2016. (co-author, SCI IF:
6.302)
6. Chunjiang He, Hanyang Hu, Kitchener D. Wilson, Haidi Wu, Jing Feng, Siyu Xia, Jared Churko, Kun
Qu, Howard Y. Chang and Joseph C. Wu: Systematic Characterization of Long Non-Coding RNAs Reveals the
Contrasting Coordination of Cis- and Trans- Molecular Regulation in Human Fetal and Adult Heart:
CIRCGENETICS. February 2016. (SCI)
7. Limin Zhou, Wei Zheng, Majing Luo, Jing Feng, Zhichun Jin, Yan Wang, Dunlan Zhang: dbCerEx - A
web-based database for the analysis of cervical cancer transcriptomes. PLOS one. 2014 9(6): 1-5. (SCI).
1、Tumor-specific circular RNA database
Cancer-specific circular RNA screening was performed on cell lines from 19 cancers, MRE and RBP
binding sites, and ORF reading frames were predicted, and alternative splicing of the parental genes was
defined.
http://gb.whu.edu.cn/CSCD
Associate Researcher, School of Computer science, Wuhan University, mainly engaged in bioinformatics, artificial intelligence and big data research, mainly focusing on optimization theory, pattern recognition, statistical machine learning and other methods, carried out single cell biomedical big data integration mining analysis model and algorithm research.Her main achievements have been published in Genome Biology, Nucleic Acids Research, IEEE Transactions on Fuzzy Systems, etc
September 2013 to June 2018, Master and Doctoral degree at Academy of Mathematics and Systems Science,
Chinese Academy of Sciences;
From August 2018 to July 2021, worked as a postdoctoral fellow at University of California, Irvine,
USA;
In September 2021, joined the School of Computer Science, Wuhan University
1.Lihua Zhang, Qing Nie. scMC learns biological variation through the alignment of
multiple single cell
genomics datasets. Genome Biology. 2021, 22(1):10.
2.Suoqin Jin*,Lihua Zhang*(co-first author), Qing Nie. scAI: an unsupervised approach for
integrative analysis of parallel single cell transcriptomic and epigenomic profiles. Genome
Biology.2020, 21(1):25.
3.Lihua Zhang, Shihua Zhang. Learning common and specific patterns from data of
multiple interrelated biological scenarios with matrix factorization. Nucleic Acids
Research.
2019, 47(13): 6606-6617.
4.Lihua Zhang, Shihua Zhang. A general joint matrix factorization framework for data
integration and its systematic algorithmic exploration. IEEE Transactions on Fuzzy
Systems. 2019, 28(9): 1971-1983.
5.Lihua Zhang,Shihua Zhang. Imputing single-cell RNA-seq data by considering cell
heterogeneity and prior expression of dropouts. Journal of Molecular Cell Biology.
2020, doi:10.1093/jmcb/mjaa052.
6.Lihua Zhang, Shihua Zhang. Comparison of computational methods for imputing
single-cell RNA-sequencing data.IEEE/ACM Transactions on Computational Biology and
Bioinformatics. 2020, 17(2): 376-389.
7.Suoqin Jin, Christian Guerrero-Juarez,Lihua Zhang, Ivan Chang, Raul Ramos,
Chen-Hsiang Kuan, Peggy Myung, Maksim V. Plikus, Qing Nie. Inference and analysis of cell-cell
communication using CellChat. Nature Communications. 2021, 12(1088).
8.Laiyi Fu, Lihua Zhang, Emmanuel Dollinger, Qinke Peng, Qing Nie#, Xiaohui Xie#.
Predicting transcription factor binding in single cells through deep learning. Science
Advances. 2020, 6(51): eaba9031.
Mainly engaged in single-cell biomedical big data mining and machine learning model and algorithm
research, and works closely with biomedical scientists to solve cutting-edge scientific
problems.Specifically, it includes the following four aspects: 1.
Single cell transcriptome, epigenome and other multi-omics data mining and integration modeling and
algorithm design
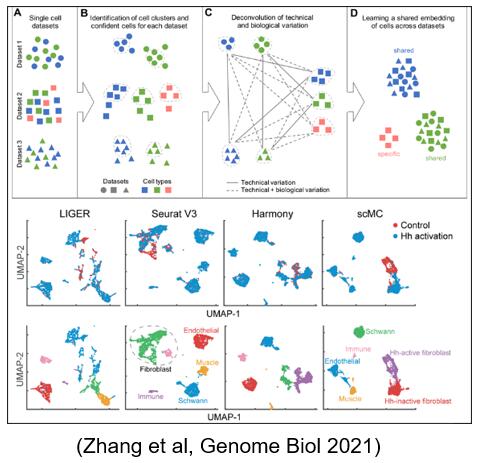
Based on analysis of variance and optimization technology, an optimization model and algorithm scMC for integrating single-cell transcriptome or epigenome biological data under various conditions are proposed. It is proposed for the first time to prevent data over integration by distinguishing biological and technical noise. Compared with the mainstream single-cell data integration methods Seurat, harmony and LIGER, scMC can integrate single-cell data more accurately and mine potential common and different cell LI subsets, especially the data from different biological conditions.
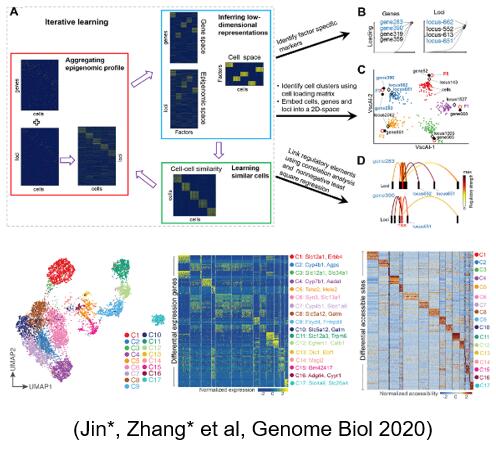
Aiming at the sparse and binary characteristics of single-cell epigenome data, a method of adaptive sparse signal enhancement based on optimization technology is innovatively proposed scAI, which is integrated with single-cell transcriptome data through iterative learning framework, dimensionality reduction and mining analysis, which provides an efficient calculation method for the integration and analysis of single-cell multiomics data. Using scAI, it was revealed that there were significant differences in chromatin opening between cell subsets with similar gene expression profiles.
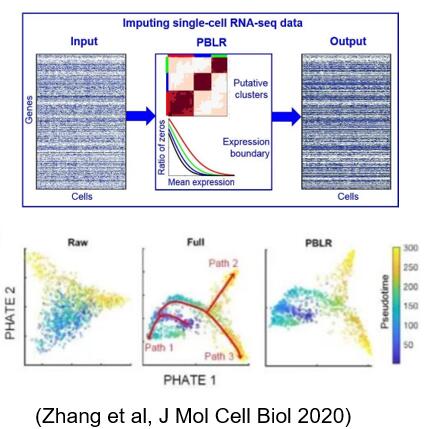
Taking full advantage of the distribution characteristics of single-cell transcriptome data, a single-cell transcriptome sequencing data filling method PBLR based on the prior information of cell subgroup and gene expression deletion is proposed, which can effectively solve the problem of data deletion and sparsity and improve the accuracy of single-cell transcriptome data analysis. Compared with the six mainstream single-cell data filling methods, PBLR can recover the missing data more accurately, and can effectively improve the downstream analysis ability of data low-dimensional visualization and extracting the relationship between genes.
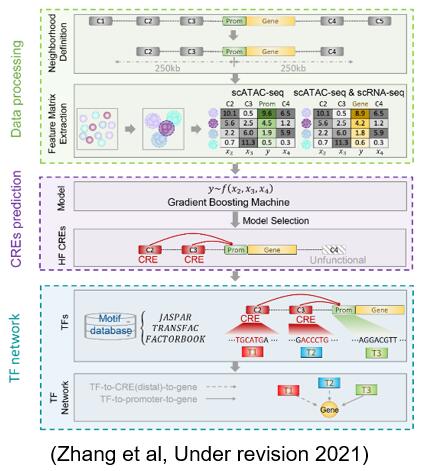
The development of single cell chromatin open capture technology provides an opportunity to study the transcriptional regulation mechanism at the single cell level. It is proposed to integrate single-cell transcriptome and chromatin openness data, identify highly functional transcriptional regulatory elements, and construct a toolkit of transcriptional regulatory elements specific to cell subsets. By this method, the differential regulatory elements and their regulatory networks corresponding to the two cell subtypes in the brain were found, and their relationship with the mutant genes related to depression was pointed out.
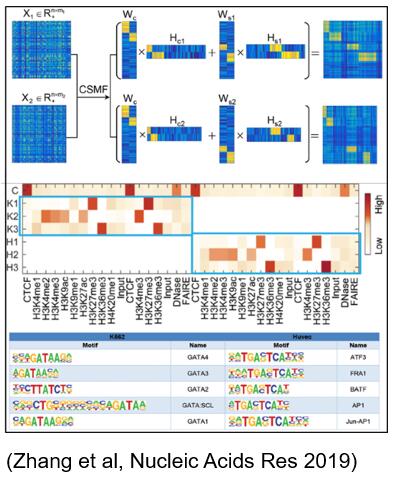
High throughput sequencing technology has produced large-scale interrelated biomedical omics data, such as different cancers and different times. How to carry out pattern recognition and difference analysis on these interrelated omics data is an important and challenging problem. The first optimization model for simultaneous identification of common and specific combination patterns is proposed. The model is based on joint nonnegative matrix decomposition technology and innovative parameter estimation method. It has strong interpretability and is easy to extract common and specific patterns with significant biological significance from the data of related biological scenarios.
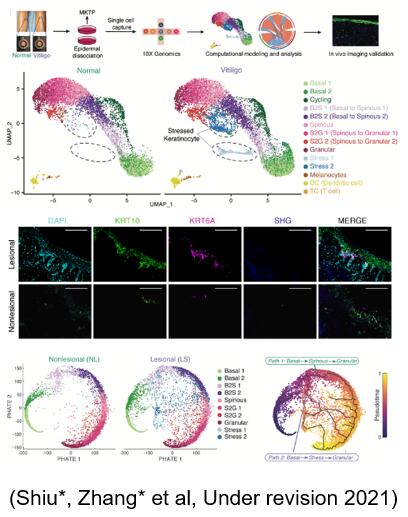
The developed single-cell data integration algorithm tool scMC, in cooperation with physicians, revealed the high heterogeneity of vitiligo and found the pathogenic signal pathway. Specific stressed keratinocytes cells were detected in vitiligo samples, which showed metabolic differences such as oxidative phosphorylation and glycolysis. In addition to the normal differentiation process, there is also a differentiation path through stress keratinocytes cells in vitiligo samples. The communication differences between keratinocytes and immune cells are found in cell interaction analysis. The results of these calculation and analysis have been further verified by experiments.